
 Il Giorno 31 gennaio 2019 alle ore 14.00 presso l’Aula Magna “De Benedictis” AOU Policlino di Bari si è tenuto un importante Convegno riguardante gli “Aggiornamenti sulle Procedure Trapiantologiche in Ematologia”. Sono intervenuti vari Clinici del settore: il Dr G. Migliore, il Dr L. Gesualdo, la Prof.ssa G. Specchia, il Dr N. Santoro,, il Prof. F. Locatelli, la Dott.ssa F. Del Bufalo, il Prof. G. La Nasa.
Il Giorno 31 gennaio 2019 alle ore 14.00 presso l’Aula Magna “De Benedictis” AOU Policlino di Bari si è tenuto un importante Convegno riguardante gli “Aggiornamenti sulle Procedure Trapiantologiche in Ematologia”. Sono intervenuti vari Clinici del settore: il Dr G. Migliore, il Dr L. Gesualdo, la Prof.ssa G. Specchia, il Dr N. Santoro,, il Prof. F. Locatelli, la Dott.ssa F. Del Bufalo, il Prof. G. La Nasa.
Erano presenti, in rappresentanza del mondo della Talassemia, di UNITED e come Associazioni Pugliesi: l’Associazione Thalassemici Brindisi, l’Associazione Bambino Thalassemico Taranto ONLUS, l’Associazione Italiana Thalassemici sez. prov.le Bari – ONLUS.
 Dopo i saluti istituzionali del Dr. G. Migliore (Direttore Generale AOU Policlino Bari) e del Dr L. Gesualdo (Presidente Scuola Medicina Bari), i ringraziamenti e l’introduzione ai lavori della Prof.ssa G. Specchia (UOC Ematologia con Trapianto Policlinico Bari) e del Dr. N. Santoro (UOC Pediatria ad Ind. Oncoematologico Policlinico Bari), il primo intervento è stato quello sulle “Complicanze precoci e tardive nel trapianto di cellule staminali emapoietiche” da parte del Prof. F. Locatelli (Direttore Dip. Ematologia e Oncologia Pediatrica IRCCS Bambino Gesù Roma).
Dopo i saluti istituzionali del Dr. G. Migliore (Direttore Generale AOU Policlino Bari) e del Dr L. Gesualdo (Presidente Scuola Medicina Bari), i ringraziamenti e l’introduzione ai lavori della Prof.ssa G. Specchia (UOC Ematologia con Trapianto Policlinico Bari) e del Dr. N. Santoro (UOC Pediatria ad Ind. Oncoematologico Policlinico Bari), il primo intervento è stato quello sulle “Complicanze precoci e tardive nel trapianto di cellule staminali emapoietiche” da parte del Prof. F. Locatelli (Direttore Dip. Ematologia e Oncologia Pediatrica IRCCS Bambino Gesù Roma).
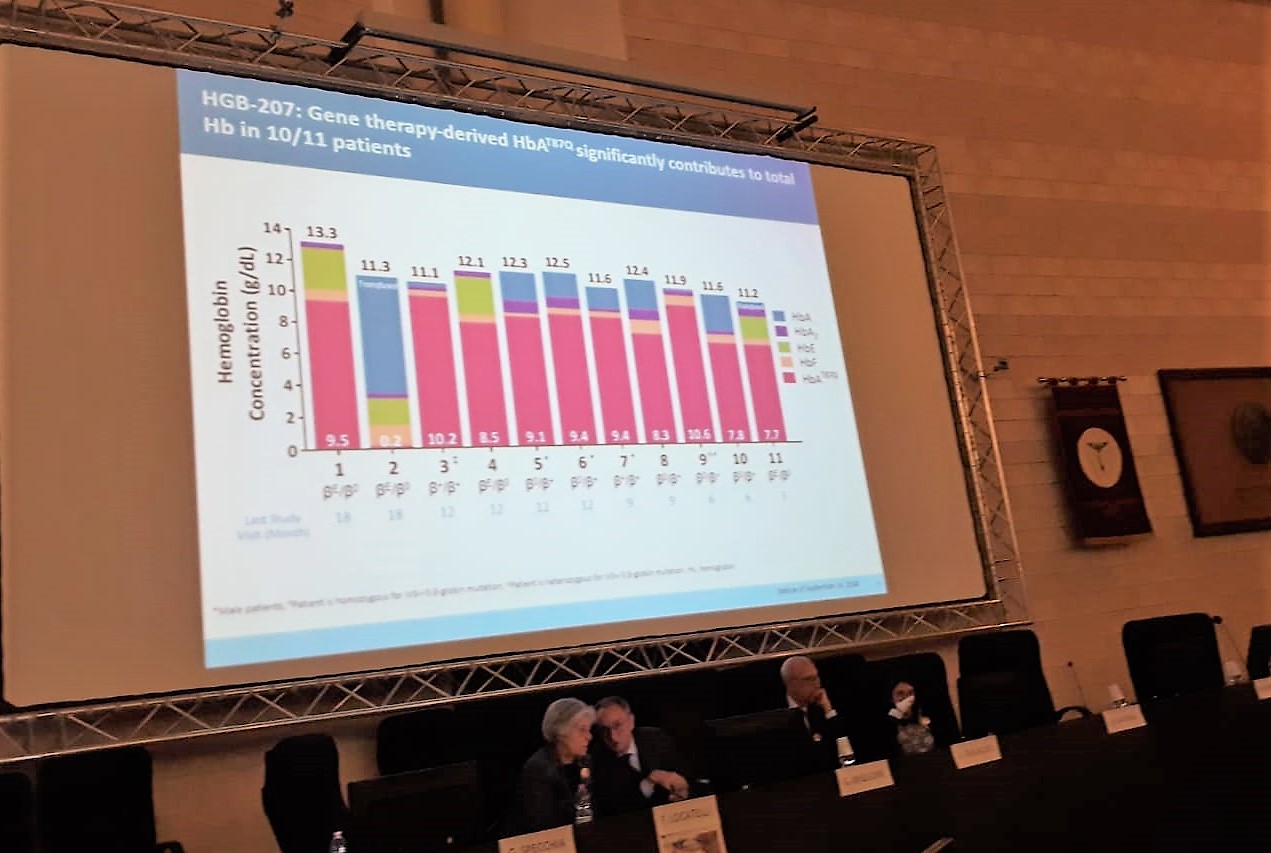
Il Professore illustra in maniera chiara e minuziosa la delicata e complicata tematica della Terapia Genica, facendo riferimento ai dati della ricerca internazionale del 60esimo Meeting Annuale svoltosi a San Diego (California) dall’1 al 4 dicembre. I nuovi dati annunciati provenienti dagli studi clinici di Fase III Northstar-2 (HGB-207) e Northstar-3 (HGB-212), condotti per valutare l’impiego Terapia Genica a base di LentiGlobin nel trattamento di pazienti con beta talassemia trasfusione dipendente (TDT) sono risultati positivi.
La Terapia Genica a base di LentiGlobin, con somministrazione una tantum, è stata sviluppata come potenziale trattamento rivolto alla disfunzione genetica che è all’origine della TDT, per ridurre o eliminare la necessità di continue trasfusioni nei pazienti. I risultati degli studi di Fase III derivano dall’analisi di un campione complessivo di 37 pazienti (pediatrici, adolescenti e adulti) affetti da TDT, sia con genotipo non β0/β0 o β0/β0, sia con mutazioni IVS-I-110, tutti trattati con LentiGlobin. Per quanto riguarda lo studio di Fase III (HGB-207) i risultati si basano sul monitoraggio dei livelli di HbA, ossia l’emoglobina sintetizzata grazie all’azione della Terapia Genica con LentiGlobin, la cui produzione determina un aumento dei livelli di emoglobina totale che ha l’obiettivo di eliminare la necessità di trasfusioni nei pazienti. Il campione dello studio è composto da 16 pazienti con genotipo non β0/β0, dei quali due pediatrici. Di questi pazienti, 11 hanno eseguito almeno 3 mesi di follow-up e 10 di loro hanno interrotto la somministrazione di trasfusioni, con un livello di emoglobina di 11.1 – 13.3 g/dL all’ultimo controllo. Inoltre, il livello di HbA in questi 10 pazienti variava tra 7.7 e 10.6 g/dL, contribuendo al 67-92% di emoglobina totale.
Un’ ulteriore indagine, basata sulla valutazione del rapporto mielo-eritroide, è stata condotta sui campioni di midollo osseo di 6 pazienti con 12 mesi di follow-up. Un basso rapporto mielo-eritroide rappresenta un aspetto chiave della diseritropoiesi, l’anomala produzione di globuli rossi nel midollo osseo che è tipica della TDT. In 5 pazienti che avevano interrotto le trasfusioni croniche si è osservato un aumento del rapporto mielo-eritroide, che suggerisce un miglioramento nella produzione di globuli rossi.
 Per quanto riguarda lo studio di Fase III (HGB-212) i risultati derivano da un campione di 3 pazienti affetti da TDT con genotipo β0/β0 o con una mutazione IVS-I-110, tutti trattati con LentiGlobin. All’ultima valutazione, tutti i pazienti, tra cui uno pediatrico, presentavano un livello di emoglobina totale maggiore di 10 g/dL. Nei due studi, il profilo di sicurezza della Terapia Genica con LentiGlobin, comprendente la comparsa di eventi avversi gravi (SAE) di patologia epatica vaso-occlusiva, è apparso coerente con il profilo di sicurezza che contraddistingue il condizionamento mieloablativo con Busulfan, generalmente eseguito prima di un trapianto di midollo osseo. Il Professore ha chiarito che dopo il trattamento con LentiGlobin, i pazienti nello studio Northstar-2 hanno iniziato a produrre emoglobina derivata dalla terapia genica, e livelli complessivi di emoglobina vicini alla norma, motivo per cui la maggior parte di loro non ha più avuto bisogno di trasfusioni. Tuttavia, l’utilizzo di queste cellule senza alcuna manipolazione rischia di causare gravi complicanze, potenzialmente fatali, correlate alla procedura trapiantologica stessa. Per questo motivo, fino a pochi anni fa, si utilizzava un metodo di “purificazione” di queste cellule che garantiva una buona percentuale di successo del trapianto (attecchimento) ma che, sfortunatamente, si associava ad un elevato rischio infettivo (soprattutto nei primi mesi dopo il trapianto) con un’elevata incidenza di mortalità. Come risultato finale, i trapianti da uno dei due genitori avevano una probabilità di successo significativamente inferiore a quella ottenibile impiegando come donatore un fratello o una sorella, o un soggetto identificato al di fuori dell’ambito familiare.
Per quanto riguarda lo studio di Fase III (HGB-212) i risultati derivano da un campione di 3 pazienti affetti da TDT con genotipo β0/β0 o con una mutazione IVS-I-110, tutti trattati con LentiGlobin. All’ultima valutazione, tutti i pazienti, tra cui uno pediatrico, presentavano un livello di emoglobina totale maggiore di 10 g/dL. Nei due studi, il profilo di sicurezza della Terapia Genica con LentiGlobin, comprendente la comparsa di eventi avversi gravi (SAE) di patologia epatica vaso-occlusiva, è apparso coerente con il profilo di sicurezza che contraddistingue il condizionamento mieloablativo con Busulfan, generalmente eseguito prima di un trapianto di midollo osseo. Il Professore ha chiarito che dopo il trattamento con LentiGlobin, i pazienti nello studio Northstar-2 hanno iniziato a produrre emoglobina derivata dalla terapia genica, e livelli complessivi di emoglobina vicini alla norma, motivo per cui la maggior parte di loro non ha più avuto bisogno di trasfusioni. Tuttavia, l’utilizzo di queste cellule senza alcuna manipolazione rischia di causare gravi complicanze, potenzialmente fatali, correlate alla procedura trapiantologica stessa. Per questo motivo, fino a pochi anni fa, si utilizzava un metodo di “purificazione” di queste cellule che garantiva una buona percentuale di successo del trapianto (attecchimento) ma che, sfortunatamente, si associava ad un elevato rischio infettivo (soprattutto nei primi mesi dopo il trapianto) con un’elevata incidenza di mortalità. Come risultato finale, i trapianti da uno dei due genitori avevano una probabilità di successo significativamente inferiore a quella ottenibile impiegando come donatore un fratello o una sorella, o un soggetto identificato al di fuori dell’ambito familiare.
Negli ultimi anni, si è messa a punto una nuova tecnica di manipolazione delle cellule staminali che permette di eliminare le cellule pericolose (linfociti T alfa/beta+), responsabili dello sviluppo di complicanze legate all’aggressione da parte di cellule del donatore sui tessuti del ricevente, lasciando però elevate quantità di cellule buone (linfociti T gamma/delta+ e cellule Natural Killer), capaci di proteggere il paziente da infezioni severe e dalla ricaduta di malattia. In particolare, il ruolo delle cellule Natural Killer da oltre 10 anni è stato oggetto di uno studio approfondito e meticoloso, che ha permesso di capire che con il nuovo approccio di manipolazione selettiva dei tessuti da trapiantare, i pazienti possono beneficiare fin da subito dell’effetto positivo. I risultati dimostrano come il rischio di mortalità da trapianto è straordinariamente basso (nell’ordine del 5%), e, conseguentemente, la probabilità di cura definitiva per questi pazienti è superiore al 70%. Inoltre, il rischio particolarmente basso di sviluppare complicanze a breve e lungo termine correlate al trapianto ottenuto grazie a questo nuovo approccio metodologico, rende questa procedura un traguardo solo pochi anni fa impensabile, e, oggi, una realtà potenzialmente applicabile a centinaia di altri bambini nel mondo.
 E´ intervenuta in un secondo momento la Dott.ssa F. Del Bufalo (IRCCS Bambino Gesù Roma) che pone l’attenzione su una delle complicanze nel setting autologo “VOD nel setting autologo del paziente pediatrico”. La Dott.ssa chiarisce che la malattia veno-occlusiva epatica (VOD epatica) è una condizione caratterizzata da una lesione tossica dei sinusoidali del fegato, che causa un’ostruzione delle piccole vene epatiche. Nei paesi in via di sviluppo, la VOD epatica si associa in particolare al regime di condizionamento utilizzato per il trapianto delle cellule staminali ematopoietiche; infatti, il 10-60% dei pazienti (a seconda del protocollo usato per il condizionamento) sviluppa questa malattia. La VOD epatica può presentarsi anche dopo la chemioterapia o la radioterapia e colpisce sia i bambini che gli adulti. Il quadro clinico si caratterizza per l’epatomegalia dolorosa, l’ittero e la ritenzione dei liquidi, che si manifesta con un aumento del peso, edemi e ascite. Può essere presente insufficienza epatica, che si manifesta con una coagulopatia ed un’encefalopatia epatica ed è comune l’insufficienza renale funzionale. Nei casi gravi, possono verificarsi gravi infezioni batteriche o insufficienze multiple d’organo. Le diagnosi differenziali si pongono nel caso del trapianto di cellule staminali ematopoietiche con le infezioni gravi e la malattia da rigetto.
E´ intervenuta in un secondo momento la Dott.ssa F. Del Bufalo (IRCCS Bambino Gesù Roma) che pone l’attenzione su una delle complicanze nel setting autologo “VOD nel setting autologo del paziente pediatrico”. La Dott.ssa chiarisce che la malattia veno-occlusiva epatica (VOD epatica) è una condizione caratterizzata da una lesione tossica dei sinusoidali del fegato, che causa un’ostruzione delle piccole vene epatiche. Nei paesi in via di sviluppo, la VOD epatica si associa in particolare al regime di condizionamento utilizzato per il trapianto delle cellule staminali ematopoietiche; infatti, il 10-60% dei pazienti (a seconda del protocollo usato per il condizionamento) sviluppa questa malattia. La VOD epatica può presentarsi anche dopo la chemioterapia o la radioterapia e colpisce sia i bambini che gli adulti. Il quadro clinico si caratterizza per l’epatomegalia dolorosa, l’ittero e la ritenzione dei liquidi, che si manifesta con un aumento del peso, edemi e ascite. Può essere presente insufficienza epatica, che si manifesta con una coagulopatia ed un’encefalopatia epatica ed è comune l’insufficienza renale funzionale. Nei casi gravi, possono verificarsi gravi infezioni batteriche o insufficienze multiple d’organo. Le diagnosi differenziali si pongono nel caso del trapianto di cellule staminali ematopoietiche con le infezioni gravi e la malattia da rigetto.
Non è disponibile un trattamento specifico. In caso di trapianto di cellule staminali ematopoietiche, il principale trattamento di profilassi è l’uso di regimi di condizionamento meno epatotossici. La gravità e il decorso della malattia variano a seconda dei pazienti e nei casi gravi, la prognosi è poco buona e il tasso di mortalità è elevato (superiore al 90%) a causa dell’insufficienza d’organo multipla.
A seguire l’intervento del Prof. G. La Nasa (Direttore Ematologia-Centro Trapianti Midollo Osseo Ospedale A. Businco Cagliari) su “Indicazioni ed outcome del trapianto nel talassemico”. Il Professore mette a confronto le terapie convenzionali con la Terapia Genica, chiarendo che, l’unica soluzione fino a qualche anno fa disponibile per eradicare in un paziente la beta talassemia era il trapianto allogenico di midollo osseo, in grado di correggere definitivamente il difetto nella produzione di emoglobina che caratterizza questi pazienti, ma, questa procedura porta con sé limitazioni e rischi. In primo luogo, l’accesso è limitato dalla disponibilità di un donatore: un donatore familiare compatibile è identificabile in meno del 25% dei pazienti, mentre un donatore compatibile può essere identificato al di fuori dell’ambito familiare al massimo in un ulteriore 30% di pazienti. Inoltre, il trapianto allogenico ha minori probabilità di successo nei pazienti adulti, rispetto a quanto osservato nei pazienti pediatrici in ragione del maggiore rischio di complicanze severe che possono determinare la morte.
Il principio della Terapia Genica consiste nell’introdurre nelle cellule malate dei pazienti una copia sana del gene difettoso. Questo processo si ottiene “infettando” le cellule del soggetto malato attraverso un virus inattivato, e, quindi, privato del suo potere dannoso per l’uomo, appartenente alla famiglia dei lentivirus. Tramite tecniche sofisticate, questo gene viene integrato nel Dna delle cellule emopoietiche proprie del paziente. Questo processo permette di ottenere, nella cellula geneticamente corretta, la sintesi della catena beta globinica, che, di conseguenza, permetterà di formare compiutamente la complessa molecola dell’emoglobina.
Secondo gli esperti la Terapia Genica si presenta come una soluzione terapeutica più sicura del trapianto allogenico di cellule emopoietiche poiché si basa sull’uso di cellule proprie del paziente. Questo consente di minimizzare i rischi della procedura e rende ragione del possibile impiego di questo tipo di terapia anche per i pazienti in età adulta. E´ una soluzione rivoluzionaria, che permetterà di migliorare notevolmente la qualità di vita dei pazienti affetti da beta talassemia trasfusione dipendente. I pazienti attualmente trattati nelle sperimentazioni cliniche in corso, anche in Italia, mostrano di essere indipendenti da trasfusione dopo un periodo di osservazione che supera, in alcuni soggetti, addirittura i 3 anni.
Il Convegno, dopo una soddisfacente discussione fra i Clinici ed i presenti, termina alle ore 17.30 ringraziando gli organizzatori, i Clinici intervenuti per la disponibilità e la collaborazione, e, tutti i presenti in Aula.
Per UNITED il Presidente “Associazione Thalassemici Brindisi”
Luana De Gioia


{kind=link}